Molecular Dynamics Simulations Guided by X-ray Scattering Signal
Keywords: Molecular Dynamics, CUDA C, GPU, X-ray Scattering
For the Github repo on driving simulations with X-ray scattering signals, check here.
For a standalone GPU-only X-ray scattering calculator, check here.
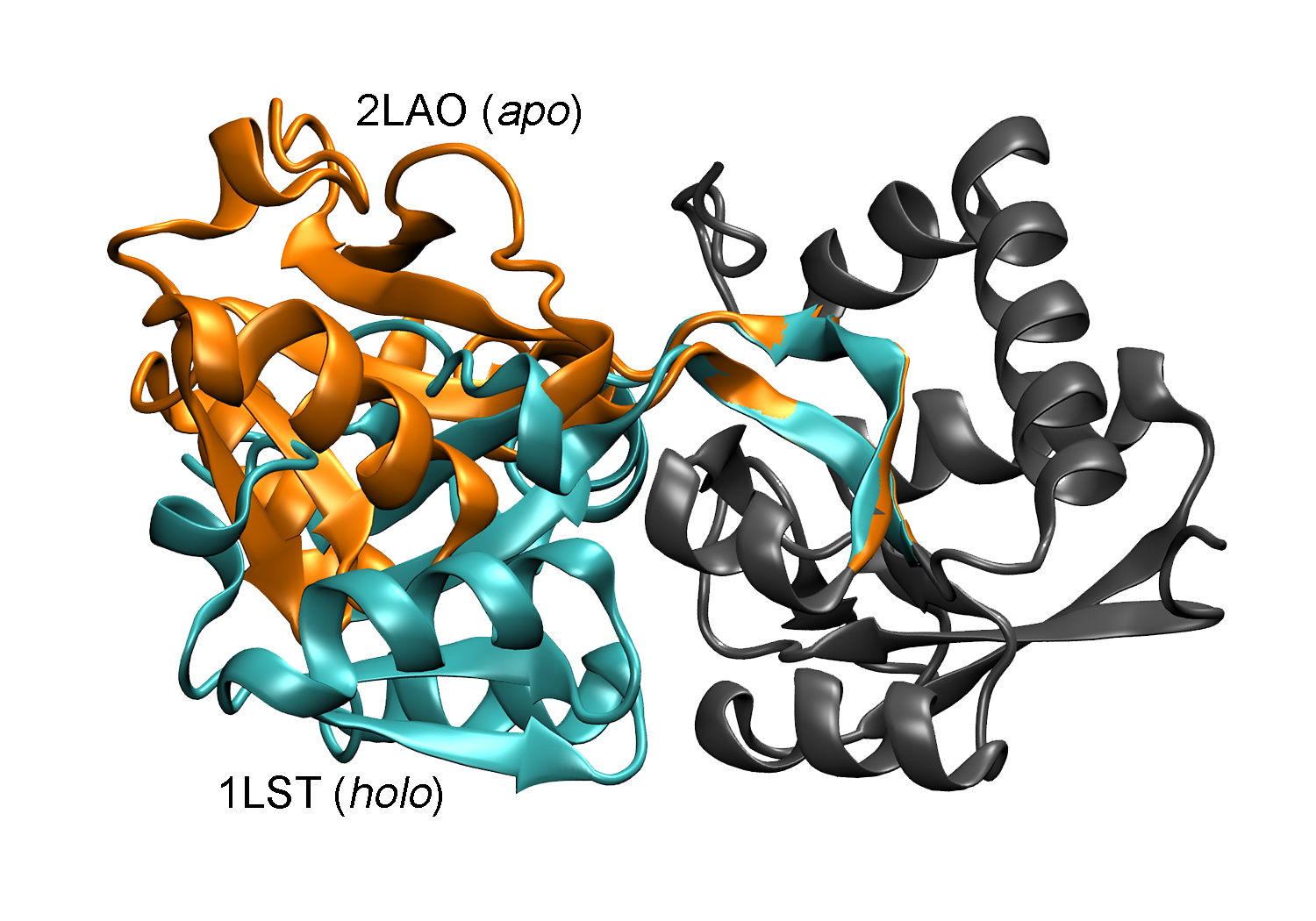
This is my main Ph.D. project where we try to guide the protein motions in molecular dynamics (MD) simulations using the data we collected, which is time-resolved X-ray solution scattering signal (TRXSS) of proteins. For example, we try to drive the 1LST structure to the 2LAO structure using only the TRXSS signal difference between the two structures.
The TRXSS signal reflects changes in the atomic distance distribution, which can be modeled with the Debye formula. However Debye formula is very expensive (proportional to number of scatterers squared) so I developed GPU code to do parallel computing, giving it a 12,000x boost in efficiency.
See our paper here: Integrating solvation shell structure in experimentally driven molecular dynamics using x-ray solution scattering data.
Finding the Unfolded State of a Protein
Keywords: Data-driven Simulation, X-ray Scattering, Molecular Dynamics
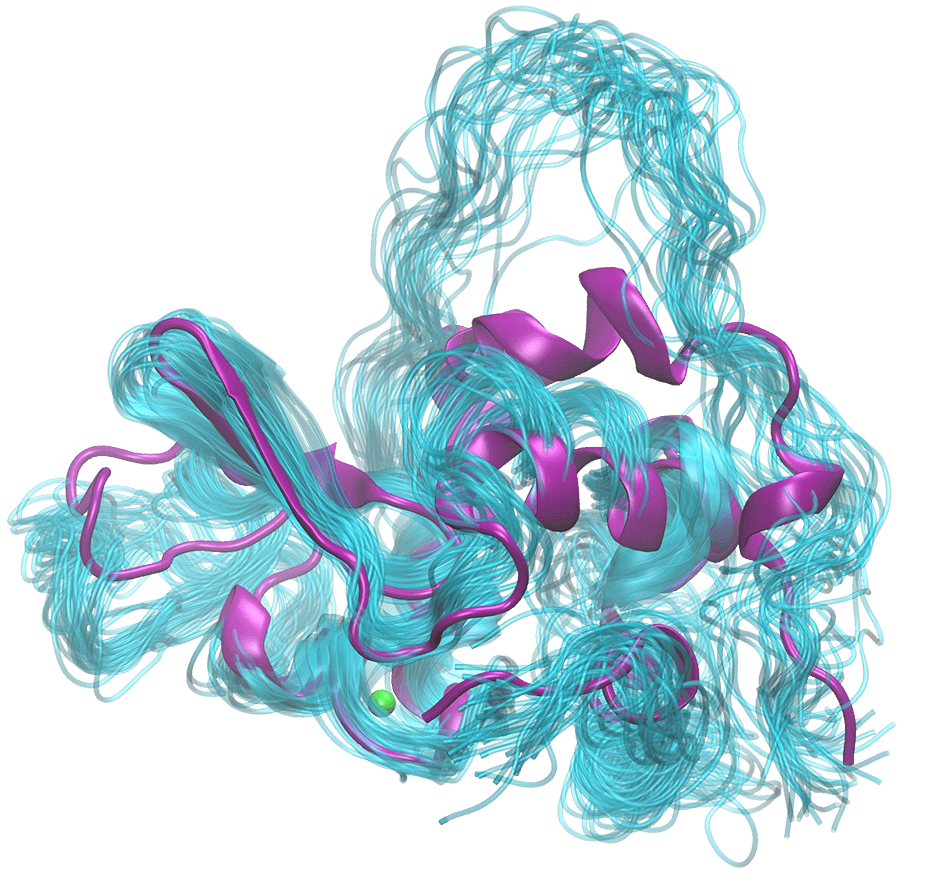
With the above method established, I decided to examine an intermediate unfolded state (molten globule) of the alpha-lactalbumin. A temperature jump is applied to the protein solution, causing the protein the unfold. We then use TRXSS to track its unfolding in real time. The signal is then input to the MD simulation, generating a different set of structures (the cyan ones in the figure; the purple one is the crystal structure) that are compatible with the data, the kinetic model, and the traditional descriptions of the molten globule state. This is then our best guess of what the protein looks like in this state, and the modeling allows us to see the structure and gain insight into the system.
See our paper here: Unfolding bovine α-lactalbumin with T-jump: Characterizing disordered intermediates via time-resolved x-ray solution scattering and molecular dynamics simulations.
High-throughput Ligand Docking Pose Refinement
Keywords: Molecular Dynamics, Machine Learning, Software Workflow
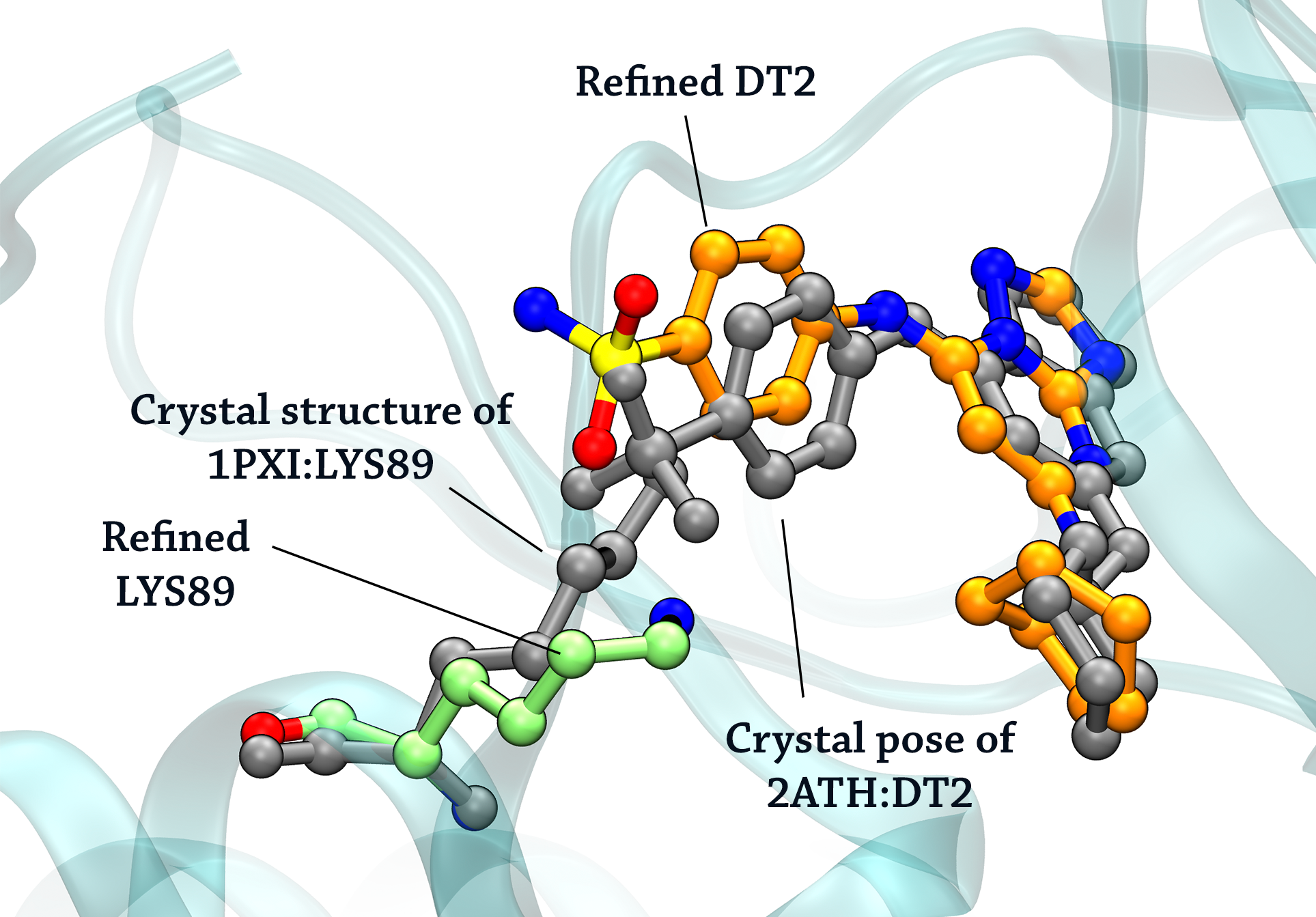
Docking pose means the output structures of the ligands generated by docking programs (e.g. AutoDock Vina) when protein is frozen. We refine this (usually low quality) prediction by allowing the ligand and the nearby protein sidechains to move in an MD fashion. Additionally we truncate to protein to form a small system, taking advantage of mdgx.cuda in the AmberTools to flood the conformational space. We also build a model zoo of XGBoost classifiers to judge which MD snapshots are more likely to be close to a crystal pose.
Deep Learning Model for Simultaneous Protein & Ligand Structure Prediction
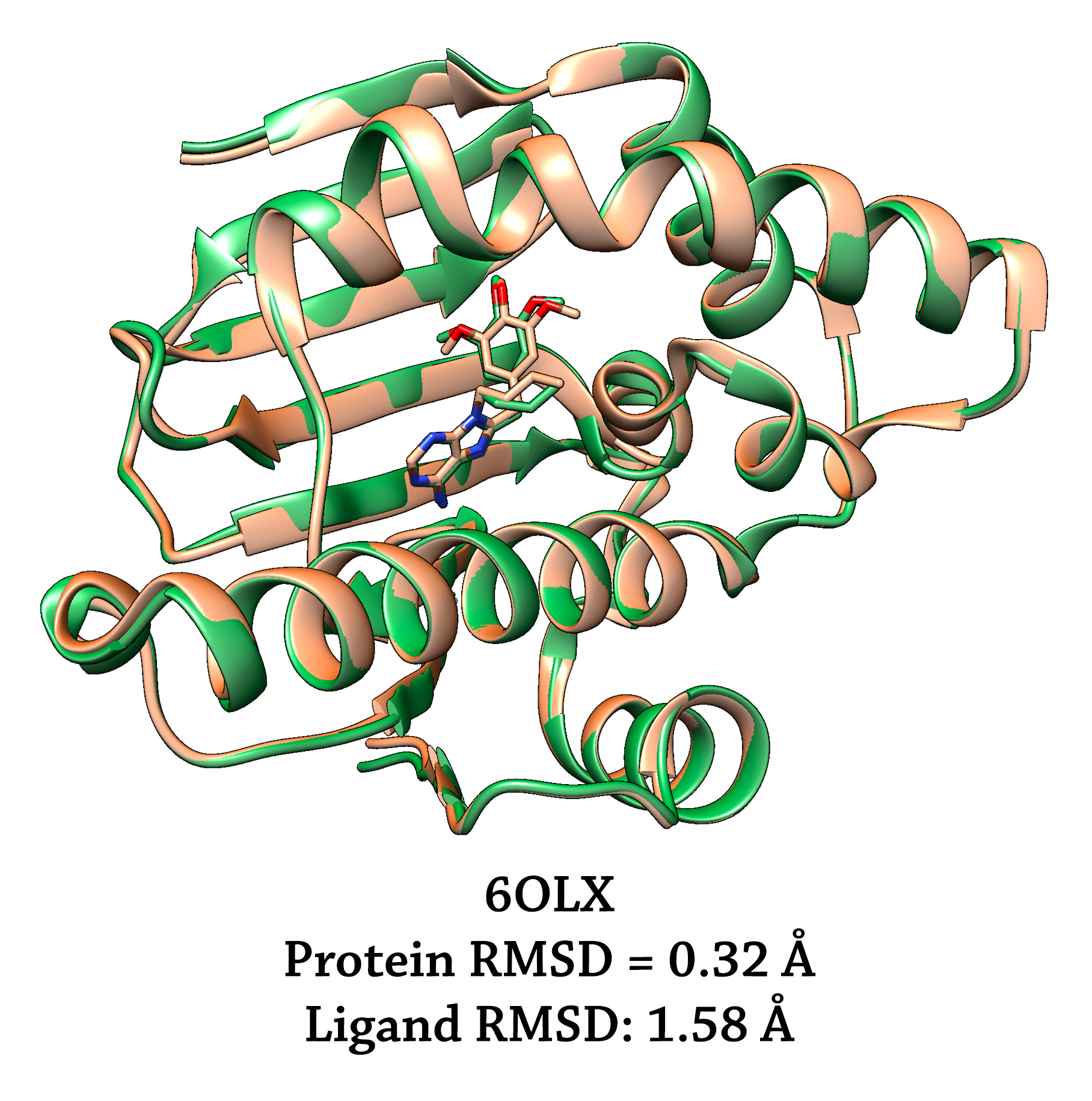
Keywords: Deep Learning, Transformers, Artificial Intelligence, Protein Folding, Ligand Binding, Structure Prediction
We built proof-of-concept, transformer-based language models to show we can predict both protein structure and ligand binding poses from just protein sequences and ligand SMILES strings. This work is a part of our TwoFold submission to the Gordon Bell Special Prize for High Performance Computing-Based COVID-19 Research, which is selected as a finalist to present in the Supercomputing 22 conference.
Side Ph.D. projects
- Designed an aluminum nitride sample holder that will not be evaporated by the IR-laser we use to generate temperature jumps.
- Probed structural dynamics of platinum and copper complexes using X-ray free electron lasers (XFEL). Multiple papers are in prep.
- Self-assembly of biomaterials as a function of temperature and pH.
- Code contribution to pytrx, a package for processing time-resolved X-ray scattering data.